Intranet
Intranet Access
Access Contact
Contact
Our analyses

Proteomics is mainly the analysis of the proteins constituting a complex protein mixture. In “bottom up” mass spectrometry, it’s the peptides resulting from an enzymatic digestion of proteins which are analyzed. In recent years, the trend has been reversed: digestion using SDS-PAGE gels has given way to digestions of proteins directly in solution.

Our facility specialized in affinity co-immunoprecipitation strategies or co-IPs.

Immunoprecipitation (IP) is a technique used to isolate a protein of interest from a cell lysate to characterize target proteins and to study protein-protein interactions.
The process requires an antibody with a high affinity and specificity for the target protein. This antibody is mixed with the sample, allowing antibody-target complexes to form. Magnetic beads that recognize these complexes are then used to “fish” and isolate the target proteins as well as any other related proteins.
When no efficient antibody is available, the target protein can be genetically modified with a peptide tag (HA, Flag, myc or GFP tags for the most common). This tag will then be targeted during the IP.

The platform performs both types of IP depending on the availability of antibodies. Protein complexes on the beads can be digested for protein identification by mass spectrometry.
A key requirement for successful immunoprecipitation analysis is the use of effective aspecificity controls.
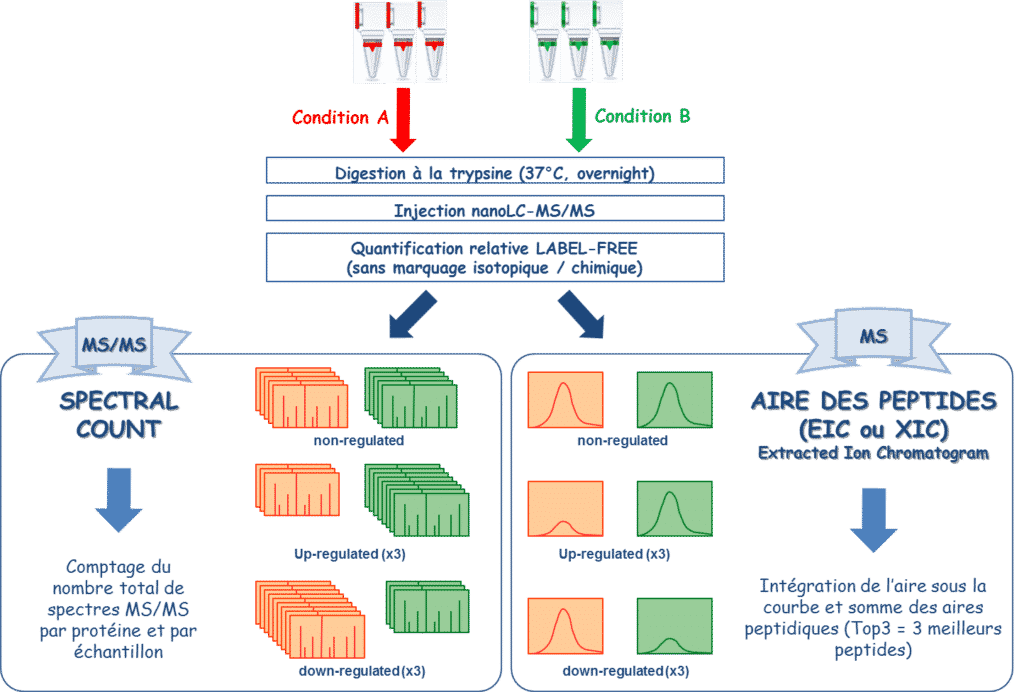
The differential proteomics approach aims to highlight proteins whose expression is modulated between two analysis conditions. This could be different treatments on the same sample, different time points … The identification of differentially expressed proteins allows the establishment of a relevant vision of the physiopathological mechanisms involved.
Label-Free analysis combines different methods for the relative quantification of proteins without isotopic labeling. Unlike quantification methods based on isotopic labelling, the samples to compare are analysed separately. The technical bias is then erased by the biological replicates (n=3 at least) and the number of conditions to be compared is no longer limited but requires the highest stability of LC and MS performances. Label-free approaches are based either on the area of detected peptides (XIC) or on the number of MS/MS spectra acquired for each peptide (spectral count).
Spectral-counting label-free analysis is the most widely used at the facility.